Accurate and equitable medical genomic analysis requires an understanding of demography and its influence on sample size and ratio
Abstract
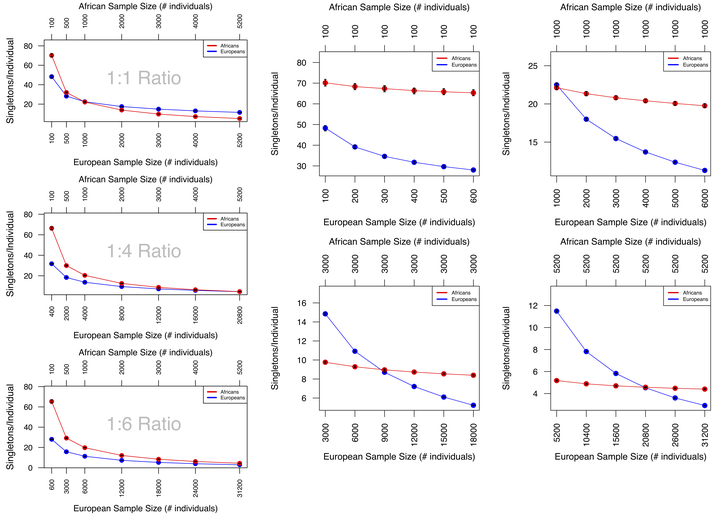
In a recent study, Petrovski and Goldstein reported that (non-Finnish) Europeans have significantly fewer nonsynonymous singletons in Online Mendelian Inheritance in Man (OMIM) disease genes compared with Africans, Latinos, South Asians, East Asians, and other unassigned non-Europeans. We use simulations of Exome Aggregation Consortium (ExAC) data to show that sample size and ratio interact to influence the number of these singletons identified in a cohort. These interactions are different across ancestries and can lead to the same number of identified singletons in both Europeans and non-Europeans without an equal number of samples. We conclude that there is a need to account for the ancestry-specific influence of demography on genomic architecture and rare variant analysis in order to address inequalities in medical genomic analysis.